惛枾壢妛愱峌偱偼丄COE僾儘僕僃僋僩偵偍偗傞惛枾壢妛偺尋媶傪岠棪傛偔恑傔傞偨傔偵丄偙偺僔儈儏儗乕僔儑儞庤朄傪庢傝擖傟丄寁嶼暔棟椞堟傪拞怱偲偡傞寁嶼僌儖乕僾偲幚尡僌儖乕僾偱嫟摨尋媶傪峴偭偰偄傑偡丅
嬶懱揑偵偼丄
|
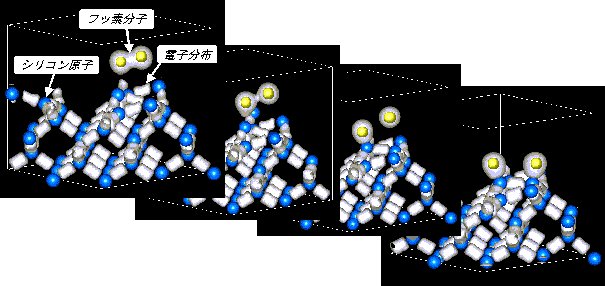
| 恾1 戞堦尨棟暘巕摦椡妛僔儈儏儗乕僔儑儞偵傛傞昞柺壔妛斀墳偺夝愅椺丅偙傟偼Si(001)2亊1昞柺忋偵偍偗傞F2暘巕偺夝棧媧拝夁掱偺寁嶼寢壥偱偁傞丅F2暘巕偑夝棧偟丄Si昞柺忋偵媧拝偡傞條巕偑尒偰庢傟傞丅偙偺傛偆偵丄戞堦尨棟暘巕摦椡妛朄偺揔梡偵傛傝條乆側壔妛斀墳夁掱偺捛愓偑壜擻偵側傞丅 |
尰嵼偺戞堦尨棟暘巕摦椡妛朄偵媮傔傜傟偰偄傞偙偲偼丄寁嶼婡幚尡傪丄幚嵺偺幚尡偵傛傝嬤偄娐嫬偺儌僨儖偱丄崅惛搙偐偮崅懍偵幚峴偡傞偙偲偑偱偒傞傾儖僑儕僘儉傪奐敪偡傞偙偲偱偡丅偲偙傠偑丄廬棃傛偔梡偄傜傟偰偄傞暯柺攇揥奐朄偵傛傞戞堦尨棟暘巕摦椡妛朄偼丄廃婜娭悢傪婎掙娭悢偵梡偄偰偄傞偨傔偵丄寁嶼儌僨儖偵惂尷傪壽偟偰偟傑偄丄尰幚偵懃偟偨儌僨儖偺峔抸偑崲擄偱偁傞偙偲偑懡乆偁傝傑偡丅偦偙偱丄摉寁嶼僌儖乕僾偱偼丄暆峀偄昞柺尰徾傪丄幚嵺偺幚尡偵傛傝嬤偄娐嫬偺儌僨儖偱僔儈儏儗乕僩偱偒傞怴偟偄寁嶼庤朄偲偟偰丄幚嬻娫嵎暘朄偵傛傞揹巕忬懺寁嶼庤朄偺奐敪偵庢傝慻傫偱偄傑偡丅偙偺曽朄偼丄儚乕僋僗僥乕僔儑儞儗儀儖偺斈梡僐儞僺儏乕僞偱傕暲楍壔偵傛傞崅懍寁嶼偑壜擻偱偁傝丄戞堦尨棟暘巕摦椡妛朄偵媮傔傜傟偰偄傞梫審傪丄慡偰枮偨偟偰偄傑偡丅尰嵼傑偱偵幚嬻娫嵎暘朄偺棟榑偺峔抸丄暲楍壔僾儘僌儔儉偺奐敪丄幚梡帋尡傪峴偄丄廬棃偺寁嶼朄偲斾妑偟偰傛傝崅惛搙側寢壥偑摼傜傟傞偙偲傪妋擣偟偰偄傑偡丅偝傜偵偼丄廬棃偺暯柺攇揥奐朄偱偼尩枾側寁嶼偑崲擄偱偁偭偨昞柺悅捈曽岦偵揹奅偑偐偐偭偨儌僨儖偺僔儈儏儗乕僔儑儞傕壜擻偱偡丅
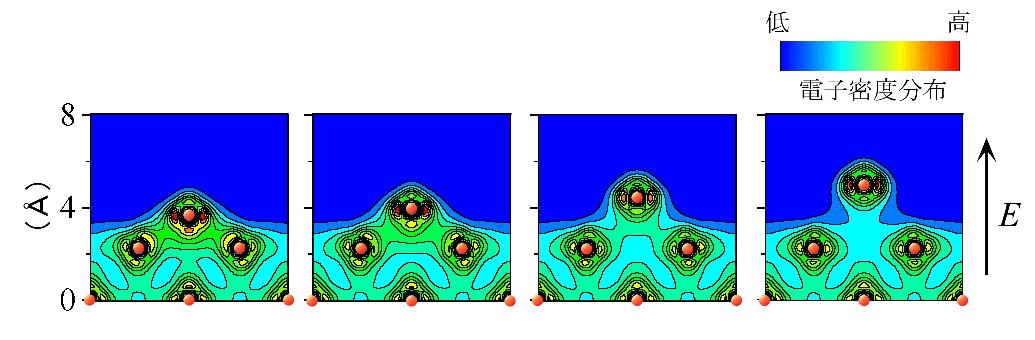 |
| 恾2 揹奅忲敪尰徾偺僔儈儏儗乕僔儑儞丅揹奅嫮搙E=6.5 (V/侌)偵偍偗傞揹巕枾搙暘晍丅1fs偼10-15昩丅揹奅偺岠壥偵傛傝媧拝偟偰偄偨僞儞僌僗僥儞尨巕偼壙揹巕傪幐偄丄梲僀僆儞偲偟偰忲敪偟偰偄偔條巕偑暘偐傞丅 |
恾2 偼僞儞僌僗僥儞(011)昞柺偐傜1屄偺僞儞僌僗僥儞媧拝尨巕偑揹奅忲敪偡傞尰徾傪僔儈儏儗乕僩偟偨傕偺偱偡丅揹奅偺岠壥偵傛傝丄僞儞僌僗僥儞媧拝尨巕偑梲僀僆儞偲側偭偰忲敪偟偰偄偔條巕偑暘偐傝傑偡丅偙偺傛偆偵怴傾儖僑儕僘儉傪丄奺庬壛岺尰徾傪巒傔偲偡傞條乆側屌懱昞柺尰徾傗壔妛斀墳偵揔梡偡傞偙偲偵傛傝丄枹抦偺昞柺尰徾偺夝柧偲墳梡媄弍偺奐敪丄摿偵敿摫懱僨僶僀僗偺惢憿媄弍偺奐敪偵峷專偱偒傞偙偲傪栚巜偟偰丄尰嵼尋媶傪恑傔偰偄傞偲偙傠偱偡丅
 |