
To enter CMD2 server, you use ssh command with your own RSA private key:
$ ssh -i .ssh/id_rsa -Y your_id@cmd2.phys.sci.osaka-u.ac.jp Enter passphrase for key '.ssh/id_rsa': ********
A compressed file of the State code is obtained from the following directory.
So let's copy (cp) and decompress (tar xzvf) the .tgz file:
$ cp /home/CMD/teac17/STATE/archives/state-cmd35.tgz . $ tar xzvf state-cmd35.tgz $ ls STATE state-cmd35.tgz
If you change directory (cd) to STATE,
you can find the following file and directories:
$ cd STATE $ ls README arch build example gncpp src util
| Directory | Description |
|---|---|
example |
Examples used in this tutorial |
gncpp |
Pseudopotential files |
src |
Source codes of State |
util |
Utility scripts |
The examples used in this tutorial are contained in example directory:
$ cd example $ ls Al C2H4 C2H4_FTMD CLEANUP CO ClonAl100 Mg Na Ni README Si
Directory Na initilly contains the following directory and files:
$ cd Na $ ls References nfinp_1 qsub.sh
| File or directory | Description |
|---|---|
References |
Directory containing output files as a reference |
nfinp_1 |
Input file |
qsub.sh |
Job script file |
The input file nfinp_1 reads:
$ cat nfinp_1
0 0 0 0 0 0
4.00 10.00 1 1 1 : GMAX GMAXP NTYP NATM NATM2
229 1 : NUM_SPACE_GROUP TYPE_BRAVAIS_LATTICE
7.90 7.90 7.90 90.00 90.00 90.00 : A B C ALPHA BETA GAMMA
12 12 12 1 1 1
0 0
0.00 0.00 0.00 1 0 1
11 0.50 22.99 6 1 0.2 : IATOMN ALFA AMION ILOC IVAN
0 0 0 0 0 0
0 1
20 20 0 84200.00 0
6 1
0 20 0.60
0.20 0.30 0.20 0.20 0.20
300.00 4 1 0.50D-09 : DTIO IMDALG IEXPL EDELTA
-10.0002 0.50D+03 0
ggapbe 1
2.00
101
4 4 4
4 4 4
6
1
0
2
0
0
0 0.00
| Parameter | Value | Description |
|---|---|---|
NTYP |
1 |
Number of atomic species |
NATM |
1 |
Number of atoms |
NATM2 |
1 |
Number of atoms (= NATM) |
NUM SPACE GROUP |
229 |
Space group number (refer to e.g. this page) |
BRAVAIS LATTICE |
1 |
Body-centered cubic |
A |
7.90 |
Length of lattice vector $\boldsymbol{A}$ in Bohr |
B |
7.90 |
Length of lattice vector $\boldsymbol{B}$ in Bohr |
C |
7.90 |
Length of lattice vector $\boldsymbol{C}$ in Bohr |
ALPHA |
90.0 |
Angle between $\boldsymbol{B}$ and $\boldsymbol{C}$ in degree |
BETA |
90.0 |
Angle between $\boldsymbol{C}$ and $\boldsymbol{A}$ in degree |
GAMMA |
90.0 |
Angle between $\boldsymbol{A}$ and $\boldsymbol{B}$ in degree |
IATOMN |
11 |
Atomic number |
EDELTA |
0.50D-09 |
Energy threshold for convergence in Hartree |
The job script is submitted using qsub command:
$ qsub qsub.csh
Your job 85 ("Na") has been submitted
If the calculation is finished successfully, some files are created in the current directory:
$ ls Na.e85 Na.pe85 References dos.data nfinp_1 nfstop.data qsub.sh Na.o85 Na.po85 STATE fort.37 nfout_1 potential.data zaj.data
| File | Description |
|---|---|
dos.data |
Density of states (DOS) |
nfstop.data |
The job is stopped if 1 is written (i.e. cat 1 > nfstop.data) |
nfout_1 |
Output file |
potential.data |
Effective potential or charge density |
zaj.data |
Wave functions |
The convergence of total energy can be seen by extracting lines including "ETOT:" from nfout_1:
$ grep ETOT: nfout_1 ETOT: 1 -0.70836409 0.7084E+00 0.1446E-03 ETOT: 2 -0.88173590 0.1734E+00 0.4949E-03 ETOT: 3 -0.89514126 0.1341E-01 0.2566E-03 ETOT: 4 -0.89514200 0.7358E-06 0.4164E-04 ETOT: 5 -0.89514201 0.1375E-07 0.7103E-05 ETOT: 6 -0.89514201 0.1805E-08 0.5763E-06 ETOT: 7 -0.89514201 0.1652E-10 0.6293E-07 ETOT: 8 -0.89514201 0.2537E-11 0.4071E-07 ETOT: 9 -0.89514201 0.1709E-11 0.5460E-08
The total energy has converged to $-0.89514201$ Hartree after nine iterations,
when energy fluctuation $0.1709\times10^{-11}$ Hartree falls below EDELTA.
To take a glance at dos.data, it is convenient to use command head or tail,
which shows only the first or last ten lines:
$ head dos.data
-13.5948134416 0.0000000000 0.0000000000
-13.5839288832 0.0000000000 0.0000000000
-13.5730443248 0.0000000000 0.0000000000
-13.5621597664 0.0000000000 0.0000000000
-13.5512752080 0.0000000000 0.0000000000
-13.5403906496 0.0000000000 0.0000000000
-13.5295060912 0.0000000000 0.0000000000
-13.5186215328 0.0000000000 0.0000000000
-13.5077369744 0.0000000000 0.0000000000
-13.4968524160 0.0000000000 0.0000000000
$ tail dos.data
8.0654577744 0.4295924878 0.4718856059
8.0763423328 0.4300690051 0.4746348279
8.0872268912 0.4305320052 0.4772607389
8.0981114496 0.4301968559 0.4797567493
8.1089960080 0.4295186395 0.4821169471
8.1198805664 0.4455473921 0.4843361241
8.1307651248 0.4395926656 0.4864097972
8.1416496832 0.4345516132 0.4883342254
8.1525342416 0.4342525693 0.4901064214
8.1634188000 0.4317972913 0.4917241594
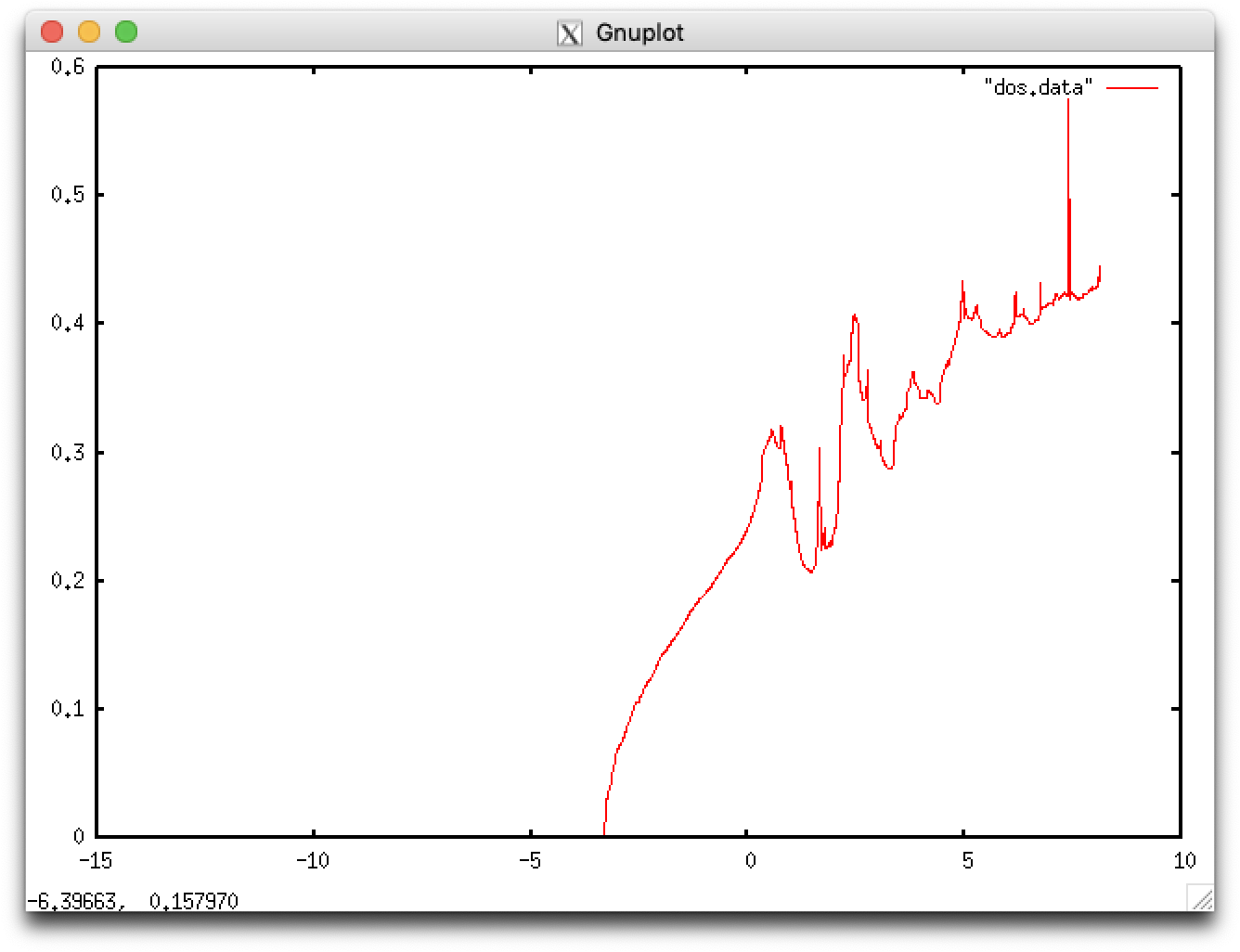
The first column corresponds to energy in eV,
while the second and third to raw and smeared data of DOS,
respectively, in eV$^{-1}$.
dos.data can be visualized by using e.g. gnuplot:
$ gnuplot
G N U P L O T
Version 4.6 patchlevel 2 last modified 2013-03-14
Build System: Linux x86_64
Copyright (C) 1986-1993, 1998, 2004, 2007-2013
Thomas Williams, Colin Kelley and many others
gnuplot home: http://www.gnuplot.info
faq, bugs, etc: type "help FAQ"
immediate help: type "help" (plot window: hit 'h')
Terminal type set to 'x11'
gnuplot> plot "dos.data" with lines

$ cd ../Mg $ cat nfinp_1
0 0 0 0 0 0
4.00 8.00 1 2 2 : GMAX GMAXP NTYP NATM NATM2
194 0 : NUM_SPACE_GROUP TYPE_BRAVAIS_LATTICE
6.10 6.10 9.60 90.0 90.0 120.0 : A B C ALPHA BELTA GAMMA
12 12 8 1 1 1
0 0 : NCORD NINV :IWEI IMDTYP ITYP
0.3333333333333 0.666666666667 0.2500000000 1 0 1
-0.3333333333333 -0.666666666667 -0.2500000000 1 0 1
12 0.20 24.31 6 1 0.0 : IATOMN ALFA AMION ILOC IVAN
0 0 0 0 0
0 1
50 50 0 36000.00 0
6 1
0 30 0.6
0.2 0.3 0.20 0.20 0.20
30.00 1 1 0.10D-9
-10.0020 0.50D+05 0
GGAPBE 1
1.00 3
101
2 2 2
2 2 2
8
1
0
2
0
0
0 0.0
| Parameter | Value | Description |
|---|---|---|
NATM |
2 |
Two Mg atoms in a primirive cell |
NUM SPACE GROUP |
194 |
Space group number of $P6_3/mmc$ |
BRAVAIS LATTICE |
0 |
Simple cubic |
NCORD |
0 |
Coodinates in units of lattice vectors |
IATOMN |
12 |
Atomic number of Mg |
$ qsub qsub.sh
Your job 87 ("Mg") has been submitted
$ grep ETOT: nfout_1 ETOT: 1 -11.69218799 0.1169E+02 0.4000E-02 ETOT: 2 -12.57326963 0.8811E+00 0.1936E-02 ETOT: 3 -12.58097106 0.7701E-02 0.9814E-03 ETOT: 4 -12.58105558 0.8451E-04 0.6903E-04 ETOT: 5 -12.58105821 0.2634E-05 0.1438E-04 ETOT: 6 -12.58105817 0.4680E-07 0.1344E-05 ETOT: 7 -12.58105814 0.2291E-07 0.4425E-06 ETOT: 8 -12.58105814 0.2388E-08 0.3259E-07 ETOT: 9 -12.58105814 0.1666E-09 0.5973E-08 ETOT: 10 -12.58105814 0.1011E-09 0.1165E-08 ETOT: 11 -12.58105814 0.1202E-10 0.2957E-09
$ gnuplot gnuplot> plot "dos.data" with lines
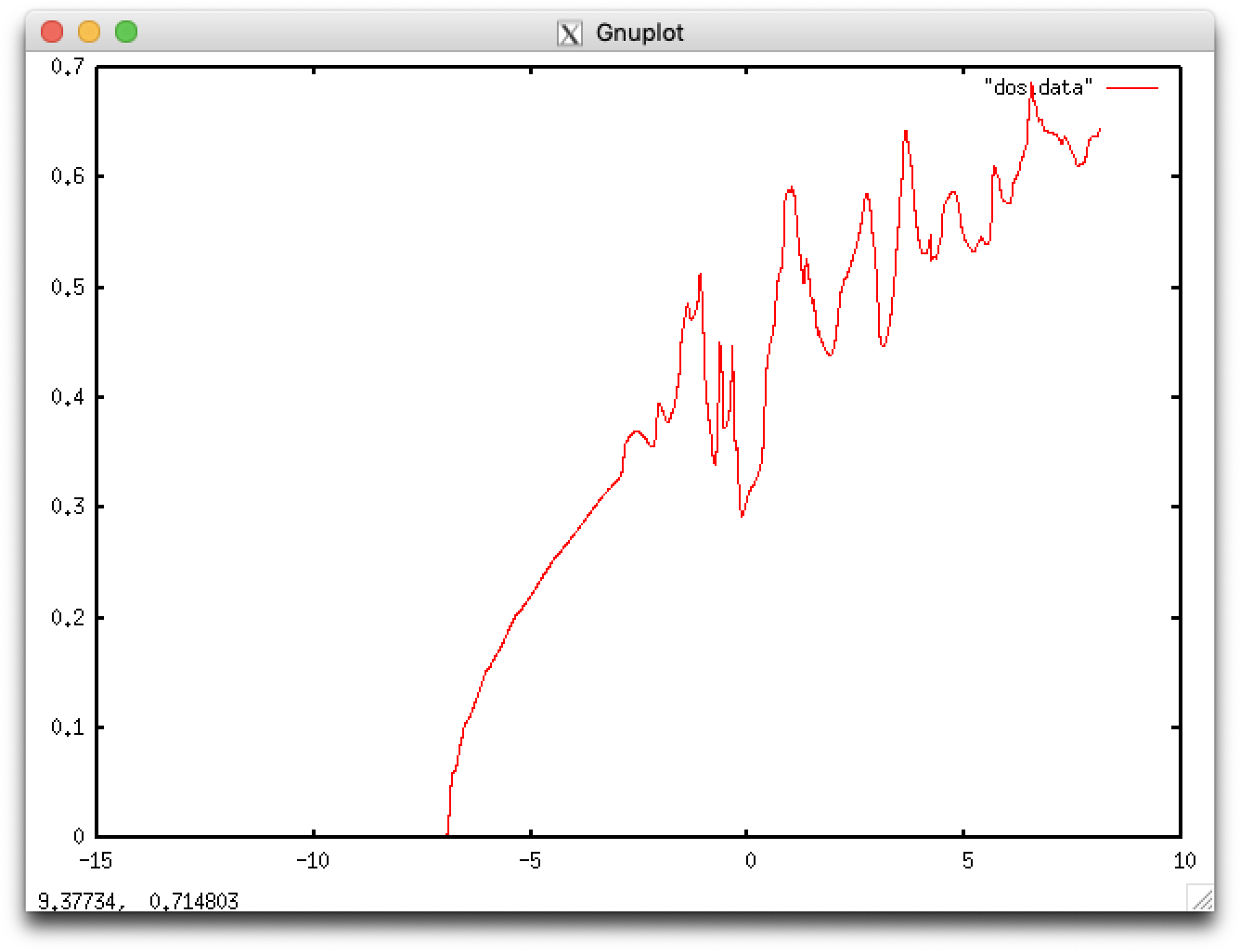
We here learn how to optimize the lattice constants of solids.

$ cd ../Al $ cat nfinp_1
0 0 0 0 0 0
4.00 8.00 1 1 1 : GMAX GMAXP NTYP NATM NATM2
221 2 : NUM_SPACE_GROUP TYPE_BRAVAIS_LATTICE
7.60 7.60 7.60 90.00 90.00 90.00 : A B C ALPHA BETA GAMMA
12 12 12 1 1 1
0 0
0.00 0.00 0.00 1 0 1
13 0.50 26.98 6 1 0.20 : IATOMN ALFA AMION ILOC IVAN
0 0 0 0 0
0 1
30 30 0 84200.00 0
6 1
0 20 0.60
0.20 0.30 0.20 0.20 0.20
300.00 4 1 0.50D-09
-0.0020 0.50D+03 0
ggapbe 1
2.00
101
4 4 4
4 4 4
6
1
0
2
0
0
0 0.00
| Parameter | Value | Description |
|---|---|---|
NATM |
1 |
One Al atoms in a primirive unit cell |
NUM SPACE GROUP |
221 (225) |
Space group number of $Pm\overline{3}m$ ($Fm\overline{3}m$) |
BRAVAIS LATTICE |
2 |
Face-centered cubic |
IATOMN |
13 |
Atomic number of Al |
We vary lattice constants A, B and C from 7.5 Bohr to 7.9 Bohr and compare calculated energies to see at which value the system is most stabilized.
The input files for repective cases are:
$ ls nfinp_*.* nfinp_7.5 nfinp_7.6 nfinp_7.7 nfinp_7.8 nfinp_7.9
The lattice constants for these input files can be seen by extracting (grep) lines including a,b,c:
$ grep "a,b,c" nfinp_*.* nfinp_7.5:7.5 7.5 7.5 90.000 90.000 90.000 : a,b,c,alpha,beta,gamma nfinp_7.6:7.6 7.6 7.6 90.000 90.000 90.000 : a,b,c,alpha,beta,gamma nfinp_7.7: 7.700000 7.700000 7.700000 90.000 90.000 90.000 : a,b,c,alpha,beta,gamma nfinp_7.8:7.8 7.8 7.8 90.000 90.000 90.000 : a,b,c,alpha,beta,gamma nfinp_7.9:7.9 7.9 7.9 90.000 90.000 90.000 : a,b,c,alpha,beta,gamma
The last block in qsub.sh
for a in 7.5 7.6 7.7 7.8 7.9
do
mpirun -np $NSLOTS ./STATE < nfinp_${a} > nfout_${a}
done
means that STATE reads these input files sequentially.
$ qsub qsub.sh
Your job 202 ("Al") has been submitted
The output files for respective lattice constants are
$ ls nfout_7.* nfout_7.5 nfout_7.6 nfout_7.7 nfout_7.8 nfout_7.9
For $a=7.5$ Bohr, the total energy converges to $-2.07149828$ Hartree:
grep ETOT: nfout_7.5 ETOT: 1 -1.61454558 0.1615E+01 0.2131E-02 ETOT: 2 -2.06781187 0.4533E+00 0.3173E-02 ETOT: 3 -2.07148674 0.3675E-02 0.1291E-02 ETOT: 4 -2.07149811 0.1138E-04 0.2187E-03 ETOT: 5 -2.07149826 0.1450E-06 0.4815E-04 ETOT: 6 -2.07149828 0.2419E-07 0.1976E-05 ETOT: 7 -2.07149828 0.3317E-09 0.4737E-06 ETOT: 8 -2.07149828 0.8983E-10 0.3199E-07 ETOT: 9 -2.07149828 0.5185E-10 0.5603E-08
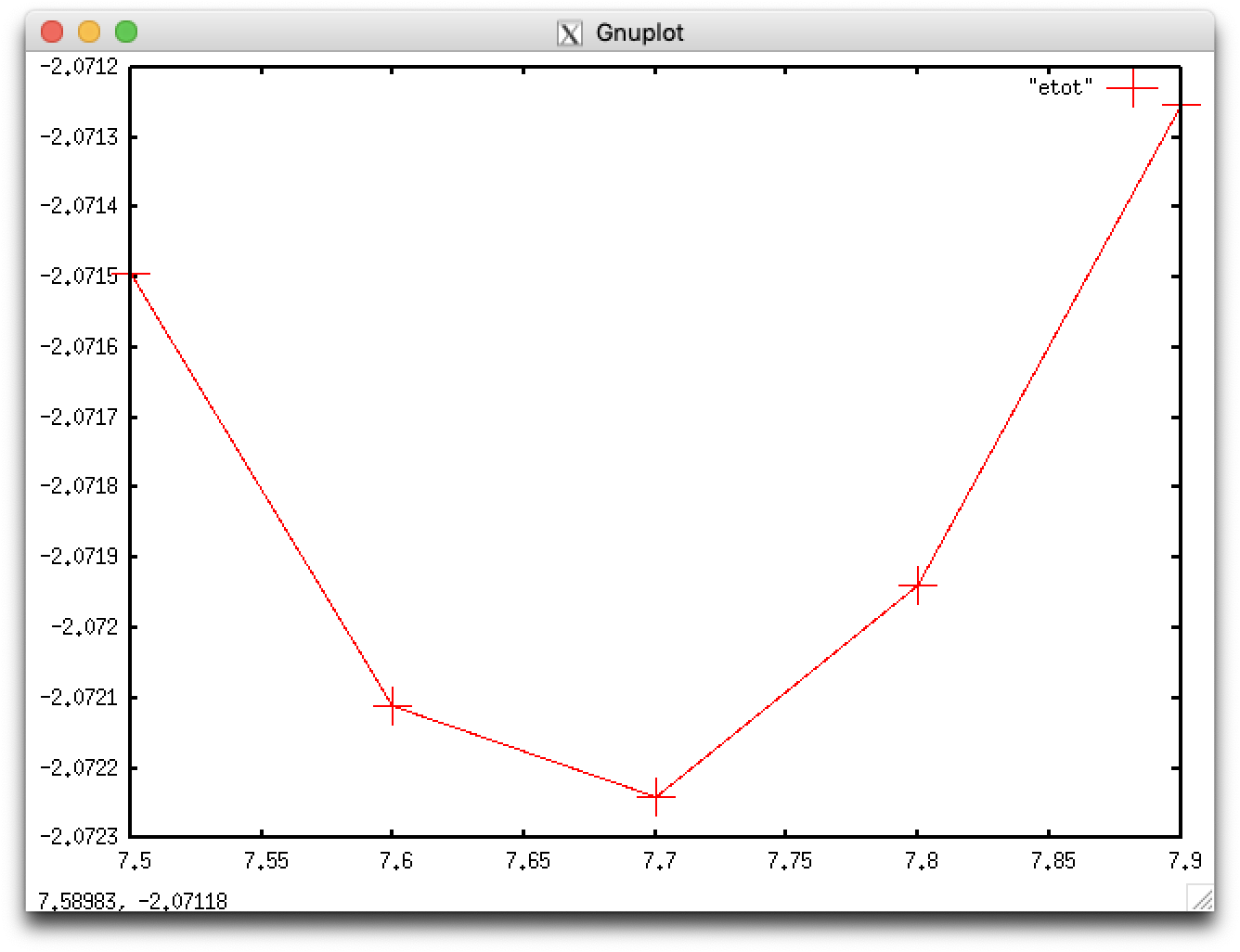
The total energies for respective lattice constants can be obtained by using script getetot.sh contained in the current directory:
$ ./getetot.sh 7.5 -2.07149508 7.6 -2.07210987 7.7 -2.07223992 7.8 -2.07193773 7.9 -2.07125357
This indicates that the lattice constans are optimized at $a\lesssim 7.7$ Bohr. Fully optimized lattice constans are obtained from least-squares fittng:
$ ./getetot.sh > etot $ gnuplot gnuplot> plot "etot" with linespoints pointsize 5
We fit the five data points with a parabolic curve \[ f(x)=ax^2+bx+c \]
gnuplot> f(x)=a*x**2+b*x+c
gnuplot> fit f(x) "etot" via a,b,c
.
.
.
Final set of parameters Asymptotic Standard Error
======================= ==========================
a = 0.0216439 +/- 0.0006142 (2.838%)
b = -0.33266 +/- 0.009459 (2.843%)
c = -0.794021 +/- 0.03641 (4.585%)
correlation matrix of the fit parameters:
a b c
a 1.000
b -1.000 1.000
c 1.000 -1.000 1.000
gnuplot> plot "etot" pointsize 5, f(x)
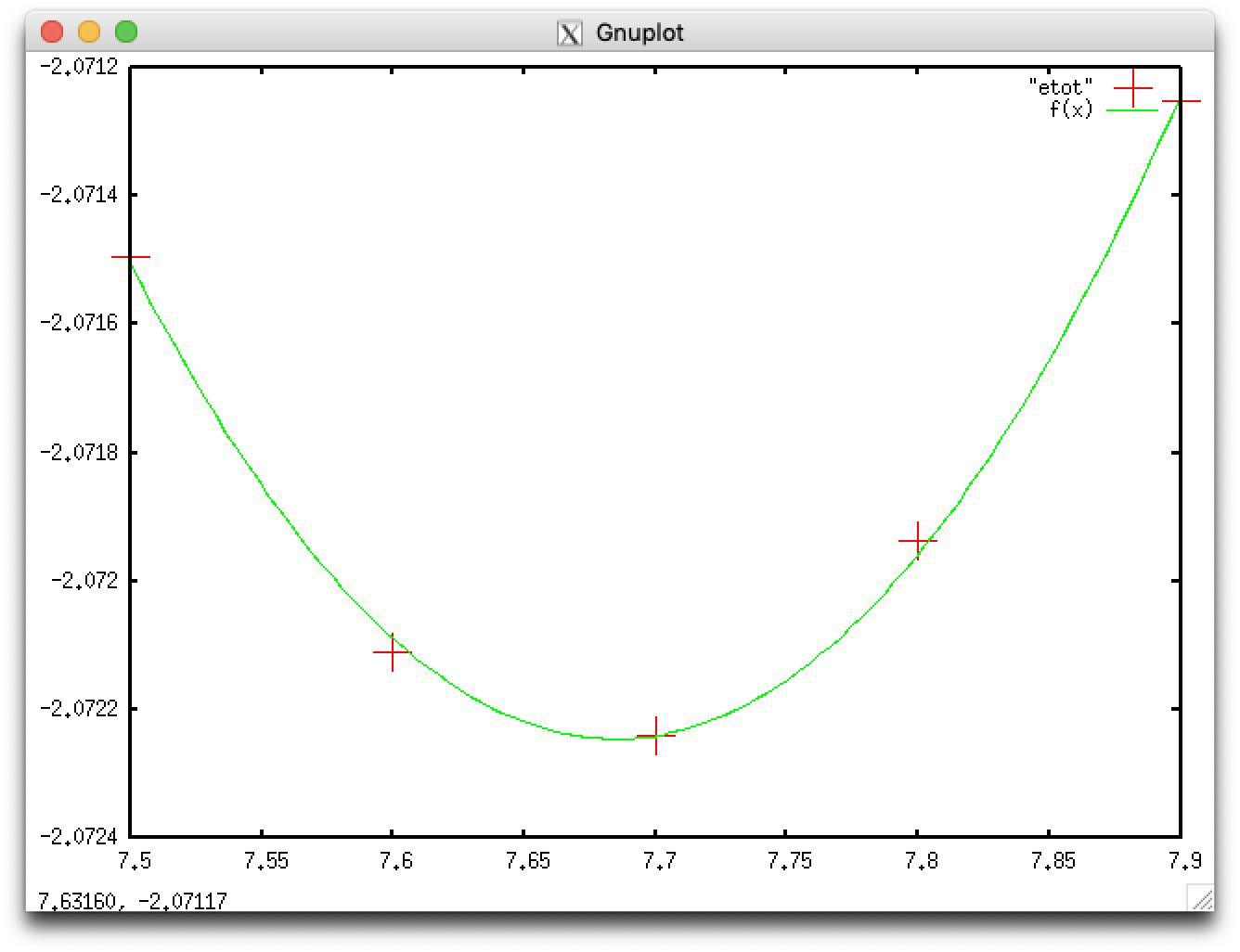
The optimal lattice constants are obtained by calculating $-\frac{b}{2a}$
$ echo "0.33266/2/0.0216439" | bc -l 7.68484422862792749920
We here consider a semiconductor with a band gap.

$ cd ../Si $ cat nfinp_1
0 0 0 0 0 0
4.00 8.00 1 2 2 : GMAX, GMAXP, NTYP, NATM, NATM2
227 2 : NUM_SPACE_GROUP TYPE_BRAVAIS_LATTICE
10.30 10.30 10.30 90.00 90.00 90.00
08 08 08 1 1 1
0 0
0.00d0 0.00d0 0.00d0 1 1 1
0.25d0 0.25d0 0.25d0 1 1 1
14 0.50 28.09 6 1 0.2 : TYPE 1IATOMN,ALFA,AMION,ILOC,IVAN
0 0 0 0 0
0 1
20 20 0 84200.00 0
6 1
0 20 0.60
0.20 0.30 0.20 0.20 0.20
300.00 4 1 0.50D-09
0.0002 0.50D+03 0
ggapbe 1
2.00
102
4 4 4
4 4 4
8
1
0
2
0
2
1
2
-15.00 5.00 0.20 501
2.400 0.20
0.20 14
0 0.00
| Parameter | Value | Description |
|---|---|---|
NATM |
2 |
Two Si atoms in a primitive cell |
NUM SPACE GROUP |
227 |
Space group number of $Fd\overline{3}m$ |
TYPE BRAVAIS LATTICE |
2 |
Face-centered cubic |
IATOMN |
14 |
Atomic number of Si |
$ qsub qsub.sh
Your job 823 ("Si") has been submitted
$ grep ETOT: nfout_1 ETOT: 1 -6.05513096 0.6055E+01 0.3203E-02 ETOT: 2 -7.84016187 0.1785E+01 0.5187E-02 ETOT: 3 -7.87270490 0.3254E-01 0.2825E-02 ETOT: 4 -7.87351715 0.8123E-03 0.6089E-03 ETOT: 5 -7.87355245 0.3530E-04 0.1887E-03 ETOT: 6 -7.87355822 0.5769E-05 0.2560E-04 ETOT: 7 -7.87355833 0.1069E-06 0.1066E-04 ETOT: 8 -7.87355833 0.4548E-08 0.1730E-05 ETOT: 9 -7.87355833 0.1916E-09 0.4194E-06 ETOT: 10 -7.87355833 0.2984E-10 0.1669E-07 ETOT: 11 -7.87355833 0.1943E-11 0.9524E-08
$ gnuplot gnuplot> plot "dos.data" with lines
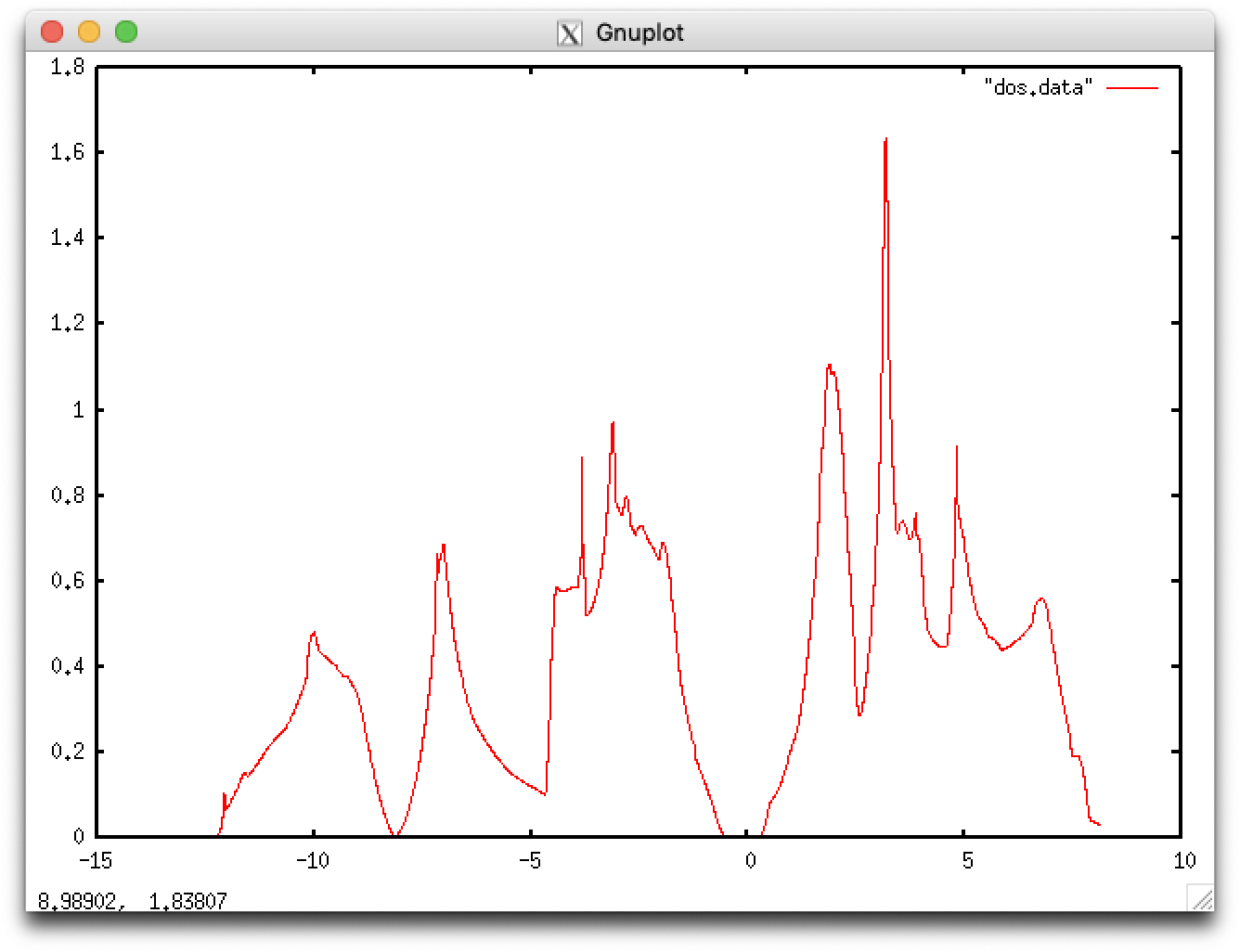
The cauclated energy gap of 0.7 eV is smaller than experimental results of 1.1 eV. Such underestimation of energy gaps is a well-known behavior of DFT results.
We here consider a ferromagnet with spin polarization.
$ cd ../Ni $ cat nfinp_1
0 0 0 0 0 0
5.00 15.00 1 1 1
221 2
6.70 6.70 6.70 90.00 90.00 90.00
12 12 12 1 1 1
0 0
0.00 0.00 0.00 1 0 1
28 0.50 58.69 6 1 0.1 : IATOMN,ALFA,AMION,ILOC,IVAN,ZETA1
0 0 0 0 0
0 1
30 30 0 84200.00 0
6 1
0 20 0.30
0.20 0.30 0.20 0.20 0.20
300.00 4 1 0.50D-09
-0.002 0.50D+03 0
ggapbe 2 : XCTYPE,KSPIN
2.00
101
4 4 4
4 4 4
10
1
0
2
0
1
1
-15.00 5.00 0.20 501
2.40000 0.200000
0.20000 14
0 0.00
| Parameter | Value | Description |
|---|---|---|
ZETA1 |
0.1 |
Initial value of relative spin polarization |
KSPIN |
2 |
Charge densities for up and down spins are treated separately |
Definition of relative spin polarization \[ \zeta=\frac{N_\uparrow-N_\downarrow}{N_\uparrow+N_\downarrow} \]
$ qsub qsub.sh
Your job 1071 ("Ni") has been submitted
$ grep ETOT: nfout_1 ETOT: 1 -48.03422182 0.4803E+02 0.1740E-01 ETOT: 2 -48.09018411 0.5596E-01 0.2913E-01 ETOT: 3 -48.35445050 0.2643E+00 0.4213E-02 ETOT: 4 -48.35581458 0.1364E-02 0.3191E-02 ETOT: 5 -48.35596078 0.1462E-03 0.2878E-02 ETOT: 6 -48.35603500 0.7422E-04 0.3500E-02 ETOT: 7 -48.35608363 0.4863E-04 0.2013E-02 ETOT: 8 -48.35608557 0.1938E-05 0.1434E-02 ETOT: 9 -48.35609022 0.4651E-05 0.3847E-03 ETOT: 10 -48.35609188 0.1668E-05 0.1860E-03 ETOT: 11 -48.35609244 0.5604E-06 0.6846E-04 ETOT: 12 -48.35609276 0.3143E-06 0.4412E-05 ETOT: 13 -48.35609276 0.2773E-08 0.4316E-05 ETOT: 14 -48.35609277 0.1732E-07 0.7494E-06 ETOT: 15 -48.35609278 0.2083E-08 0.2593E-06 ETOT: 16 -48.35609278 0.1319E-08 0.5970E-07 ETOT: 17 -48.35609278 0.1041E-09 0.3455E-07 ETOT: 18 -48.35609278 0.6774E-10 0.2829E-07 ETOT: 19 -48.35609278 0.1236E-09 0.9241E-08
Spin polarization can be confirmed by extracting lines including "Charge Density =" from nfout_1:
$ grep "Charge Density = " nfout_1 | sed -e "s/^.*=//" 5.5000000000 4.5000000000 10.0000000000 5.5000000000 4.5000000000 10.0000000000 5.0000004105 5.0000004281 9.9999991614 5.2578321672 4.7421678328 10.0000000000 5.3398749142 4.6601246078 10.0000004781 5.2975685186 4.7024314814 10.0000000000 5.2859332950 4.7140669339 9.9999997711 5.2922061685 4.7077938315 10.0000000000 5.3141836552 4.6858164843 9.9999998605 5.3013843445 4.6986156555 10.0000000000 5.3345599671 4.6654401481 9.9999998848 5.4563714243 4.5436285757 10.0000000000 5.3608062331 4.6391938548 9.9999999121 5.4119364707 4.5880635293 10.0000000000 5.3595085386 4.6404915449 9.9999999164 5.3969604451 4.6030395549 10.0000000000 5.3598549611 4.6401451225 9.9999999164 5.3636681705 4.6363318295 10.0000000000 5.3540235787 4.6459765078 9.9999999135 5.3562830503 4.6437169497 10.0000000000 5.3517383485 4.6482617376 9.9999999139 5.3518121053 4.6481878947 10.0000000000 5.3501642704 4.6498358163 9.9999999133 5.3494996637 4.6505003363 10.0000000000 5.3494117687 4.6505883184 9.9999999129 5.3495529688 4.6504470312 10.0000000000 5.3494412221 4.6505588648 9.9999999131 5.3494258883 4.6505741117 10.0000000000 5.3494045041 4.6505955829 9.9999999130 5.3494083435 4.6505916565 10.0000000000 5.3494011874 4.6505988996 9.9999999130 5.3493965623 4.6506034377 10.0000000000 5.3493981629 4.6506019240 9.9999999131 5.3493972564 4.6506027436 10.0000000000 5.3493983908 4.6506016962 9.9999999130 5.3493977310 4.6506022690 10.0000000000 5.3493985687 4.6506015183 9.9999999130 5.3493985970 4.6506014030 10.0000000000 5.3493988925 4.6506011945 9.9999999130
The first and second columns correspond to charges with up and down spins, respectively, while the third to the total charge. The initial and final values of relative spin polarization are \[ \zeta_{\rm ini}=\frac{5.50-4.50}{10.00}=0.10, \] \[ \zeta_{\rm fin}=\frac{5.34-4.65}{10.00}=0.07. \]
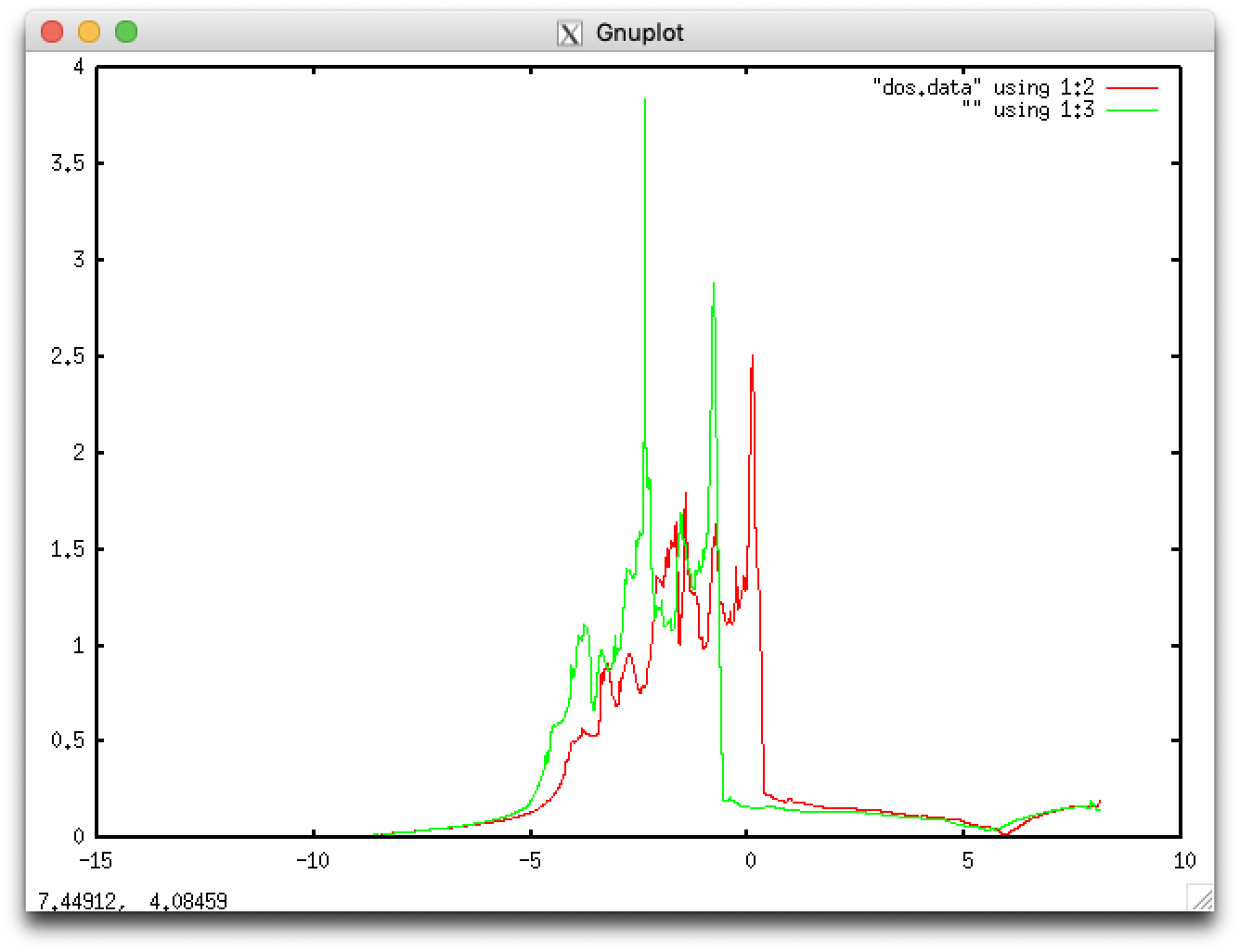
Unlike spin non-polarized calculations, dos.data consists of five columns:
$ head dos.data | tr -s " " -13.5948134416 0.0000000000 0.0000000000 0.0000000000 0.0000000000 -13.5839288832 0.0000000000 0.0000000000 0.0000000000 0.0000000000 -13.5730443248 0.0000000000 0.0000000000 0.0000000000 0.0000000000 -13.5621597664 0.0000000000 0.0000000000 0.0000000000 0.0000000000 -13.5512752080 0.0000000000 0.0000000000 0.0000000000 0.0000000000 -13.5403906496 0.0000000000 0.0000000000 0.0000000000 0.0000000000 -13.5295060912 0.0000000000 0.0000000000 0.0000000000 0.0000000000 -13.5186215328 0.0000000000 0.0000000000 0.0000000000 0.0000000000 -13.5077369744 0.0000000000 0.0000000000 0.0000000000 0.0000000000 -13.4968524160 0.0000000000 0.0000000000 0.0000000000 0.0000000000
The second and third columns correspond to raw data for up and down spins, respectively, while the fourth and fifth to smeared data for up and down spins, respectively. Spin polarization can also be confirmed by plotting the DOS data for up and down spins:
$ gnuplot gnuplot> plot "dos.data" using 1:2 with lines,"" using 1:3 with lines
We here consider a diatomic molecule in a vacuum.
Directory CO contains three input files corresponding to different diagonalization scheme:
$ cd ../CO $ ls nfinp_* nfinp_dav nfinp_rmm1 nfinp_rmm2
The last block in qsub.sh
for j in dav rmm1 rmm2
do
mpirun -np $NSLOTS ./STATE < nfinp_${j} > nfout_${j}
done
means that STATE reads these input files sequentially.
Among them, nfinp_dav reads:
$ cat nfinp_dav
0 0 0 0 0 0
5.50 20.00 2 2 2 : GMAX GMAXP NTYP NATM NATM2
1 0 : NUM_SPACE_GROUP TYPE_BRAVAIS_LATTICE
6.00 4.00 4.00 90.00 90.00 90.00
1 1 1 1 1 1
1 0 : NCORD, NINV
0.00 0.00 0.00 1 1 1 : CPS(1,1:3) IWEI IMDTYP ITYP
2.20 0.00 0.00 1 1 2 : CPS(2,1:3) IWEI IMDTYP ITYP
6 0.15 12.01 3 1 0.d0 : IATOMN ALFA AMION ILOC IVAN
8 0.15 16.00 3 1 0.d0 : IATOMN ALFA AMION ILOC IVAN
0 0 0 0 0 : ICOND INIPOS INIVEL ININOS INIACC
0 1
200 200 0 57200.00 0
3 1
0 8 0.8
0.60 0.50 0.60 0.70 1.00
30.00 2 1 0.10D-08
0.001 0.10D+02 0
ggapbe 1
1.00
102
0 0 0
0 0 0
8
1 : NEXTST
0
2 : IMSD
0
0
0 0.0
| Parameter | Value | Description |
|---|---|---|
NTYP |
2 |
Two atomic species C and O |
NATM |
2 |
One C atom and one O atom in a primitive cell |
NUM SPACE GROUP |
1 |
Space group number of $P_1$ |
TYPE BRAVAIS LATTICE |
0 |
Simple cubic |
NCORD |
1 |
Coordinates in units of Bohr |
ITYP |
1 or 2 |
Index of atomic species in the position |
The difference among the input files can be seen by using command diff:
$ diff nfinp_dav nfinp_rmm1 11c11 < 0 0 0 0 0 : ICOND INIPOS INIVEL ININOS INIACC --- > 1 0 0 0 0 : ICOND INIPOS INIVEL ININOS INIACC 25c25 < 1 : NEXTST --- > 0 : NEXTST 27c27 < 2 : IMSD --- > 1 : IMSD
$ diff nfinp_rmm1 nfinp_rmm2 27c27 < 1 : IMSD --- > 2 : IMSD
| Parameter | Value | Description |
|---|---|---|
ICOND |
0 |
Start calculation from scratch with randomly generated wave functions |
1 |
Restart calculation using old wave functinos and charge density | |
NEXTST |
0 |
Non-local pseudopotentials in the real space |
1 |
Non-local pseudopotentials in the reciprocal space | |
IMSD |
1 |
Diagonalization by means of the RMM-DIIS method (faster than the Davidson method but applicable only near convergence) |
2 |
Diagonalization by means of the Davidson method |
$ qsub qsub.sh
Your job 1265 ("CO") has been submitted
$ grep ETOT: nfout_dav ETOT: 1 -16.71058056 0.1671E+02 0.8965E-01 ETOT: 2 -20.04069483 0.3330E+01 0.6387E-01 ETOT: 3 -21.45761599 0.1417E+01 0.6830E-01 ETOT: 4 -22.15338988 0.6958E+00 0.2822E-01 ETOT: 5 -22.21588778 0.6250E-01 0.7654E-02 ETOT: 6 -22.21907373 0.3186E-02 0.1845E-02 ETOT: 7 -22.21941896 0.3452E-03 0.4914E-03 ETOT: 8 -22.21942378 0.4822E-05 0.1031E-03 ETOT: 9 -22.21942425 0.4641E-06 0.1578E-04 ETOT: 10 -22.21942426 0.9876E-08 0.4861E-05 ETOT: 11 -22.21942426 0.2909E-09 0.2236E-05 ETOT: 12 -22.21942425 0.6225E-09 0.3018E-05 ETOT: 13 -22.21942426 0.9965E-09 0.7976E-06
$ grep ETOT: nfout_rmm1 ETOT: 1 -22.21942552 0.1260E-05 0.2705E-05 ETOT: 2 -22.21942551 0.1390E-08 0.4558E-05 ETOT: 3 -22.21942551 0.4208E-08 0.7104E-05 ETOT: 4 -22.21942552 0.6704E-08 0.1284E-06 ETOT: 5 -22.21942552 0.1157E-10 0.9044E-07 ETOT: 6 -22.21942552 0.1766E-11 0.1179E-06
$ grep ETOT: nfout_rmm2 ETOT: 1 -22.21942552 0.1155E-10 0.2157E-06 ETOT: 2 -22.21942552 0.1197E-11 0.3560E-06 ETOT: 3 -22.21942552 0.4773E-10 0.5777E-06 ETOT: 4 -22.21942552 0.5626E-10 0.3069E-07
The job in qsub.sh is performed in the following order:
IMSD = 2) in the reciprocal space (NEXTST = 1)IMSD = 1) in the real space (NEXTST = 0)IMSD = 1) in the real space (NEXTST = 0)Although the total energy of the small system is fully converged in each step, it is recommended that for larger systems you follow steps 1 and 2 to accelerate convergence. To stop the first step before convergence, recall that you can stop the calculation at any monent by:
$ cat 1 > nfstop.data
We here learn how to optimize the structure of a molecule in a vacuum.
$ cd ../C2H4 $ cat nfinp_1
0 0 0 0 0 0
5.00 15.00 2 6 6
1 0
12.00 12.00 12.00 90.0 90.0 90.0
1 1 1 1 1 1
1 0
1.2627229833 0.0000000000 0.0000000000 1 1 1 : CPS(1,1:3), IWEI, IMDTYP, ITYP
2.3483288468 1.7534586685 0.0000000000 1 1 2
2.3483288468 -1.7534586685 0.0000000000 1 1 2
-1.2627229833 0.0000000000 0.0000000000 1 1 1
-2.3483288468 1.7534586685 0.0000000000 1 1 2
-2.3483288468 -1.7534586685 0.0000000000 1 1 2
6 0.15 12.010 3 1 0.d0
1 0.15 1.008 3 1 0.d0
0 0 0 0 0
0 1
100 200 0 57200.00 0
3 1
0 8 0.8
0.60 0.50 0.60 0.70 1.00
300.00 4 1 0.10D-08 : DTIO IMDALG IEXPL EDELTA
0.0010 0.05D-02 0 : WIDTH FORCCR ISTRESS
ggapbe 1
1.00 3
102
0 0 0
0 0 0
10
1
0
2
0
0
0 0.0
| Parameter | Value | Description |
|---|---|---|
IMDTYP |
1 |
Mobile atom |
EDELTA |
0.10D-08 |
Energy threshold for convergence in Hartree |
IMDALG |
4 |
Structure optimization based on the generalized direct inversion in the iterative subspace (GDIIS) method |
FORCCR |
0.05D-02 |
Force threshold for structure optimization in Hartree/Bohr |
All the atoms with IMDTYP = 1 are displaced after energy convergence
until the atomic forces fall below FORCCR.
If structure optimization is unnecessary, set FORCCR to a large value
such as 0.05D+02 (just by changing the sign)
$ qsub qsub.sh
Your job 1266 ("C2H4") has been submitted
$ grep ETOT: nfout_1 ETOT: 1 -3.16413969 0.3164E+01 0.1427E-01 ETOT: 2 -10.33933293 0.7175E+01 0.9138E-02 ETOT: 3 -13.66240712 0.3323E+01 0.9863E-02 ETOT: 4 -13.85627484 0.1939E+00 0.7894E-02 ETOT: 5 -13.87802289 0.2175E-01 0.3918E-02 ETOT: 6 -13.89907055 0.2105E-01 0.1645E-02 ETOT: 7 -13.90059984 0.1529E-02 0.8876E-03 ETOT: 8 -13.90185461 0.1255E-02 0.4448E-03 ETOT: 9 -13.90219429 0.3397E-03 0.2120E-03 ETOT: 10 -13.90229046 0.9617E-04 0.1062E-03 ETOT: 11 -13.90230633 0.1587E-04 0.5909E-04 ETOT: 12 -13.90230883 0.2499E-05 0.5240E-04 ETOT: 13 -13.90231560 0.6764E-05 0.1937E-04 ETOT: 14 -13.90231634 0.7420E-06 0.6523E-05 ETOT: 15 -13.90231644 0.1046E-06 0.2221E-05 ETOT: 16 -13.90231645 0.1107E-07 0.9719E-06 ETOT: 17 -13.90231646 0.4903E-08 0.2397E-06 ETOT: 18 -13.90231646 0.8366E-09 0.1221E-06 ETOT: 19 -13.90231646 0.3153E-09 0.7961E-07 ETOT: 1 -13.90232645 0.9985E-05 0.1217E-03 ETOT: 2 -13.90231931 0.7140E-05 0.5814E-04 ETOT: 3 -13.90233207 0.1277E-04 0.2285E-04 ETOT: 4 -13.90233243 0.3590E-06 0.6832E-05 ETOT: 5 -13.90233255 0.1137E-06 0.3583E-05 ETOT: 6 -13.90233257 0.2865E-07 0.1616E-05 ETOT: 7 -13.90233258 0.9958E-08 0.7747E-06 ETOT: 8 -13.90233259 0.7438E-08 0.4376E-06 ETOT: 9 -13.90233259 0.2115E-08 0.2911E-06 ETOT: 10 -13.90233259 0.5482E-09 0.7673E-07 ETOT: 11 -13.90233259 0.4376E-10 0.4456E-07 ETOT: 1 -13.90233554 0.2947E-05 0.8474E-04 ETOT: 2 -13.90233335 0.2195E-05 0.3483E-04 ETOT: 3 -13.90233625 0.2903E-05 0.9994E-05 ETOT: 4 -13.90233630 0.4947E-07 0.2661E-05 ETOT: 5 -13.90233633 0.3091E-07 0.1382E-05 ETOT: 6 -13.90233633 0.3928E-08 0.5785E-06 ETOT: 7 -13.90233634 0.2846E-08 0.3114E-06 ETOT: 8 -13.90233634 0.1146E-08 0.1308E-06 ETOT: 1 -13.90234254 0.6205E-05 0.6348E-04 ETOT: 2 -13.90234067 0.1870E-05 0.2972E-04 ETOT: 3 -13.90234315 0.2479E-05 0.9375E-05 ETOT: 4 -13.90234325 0.9545E-07 0.3151E-05 ETOT: 5 -13.90234327 0.1959E-07 0.1637E-05 ETOT: 6 -13.90234327 0.4244E-08 0.7358E-06 ETOT: 7 -13.90234327 0.2422E-08 0.3539E-06 ETOT: 8 -13.90234327 0.1667E-08 0.1437E-06
Every time energy converges quenched molecylar dynamics displaces atoms, and the self-consistent calculation is repeated for updated atomic configurations.
Atomic configurations and forces in $x, y$ and $z$ directions
can be seen by extracting lines including MD: from nfout_1:
$ grep MD: nfout_1 MD: 1 MD: 1 C 1.262723 0.000000 0.000000 -0.00109 0.00000 0.00000 MD: 2 H 2.348329 1.753459 0.000000 -0.00124 -0.00063 0.00000 MD: 3 H 2.348329 -1.753459 0.000000 -0.00124 0.00063 -0.00000 MD: 4 C -1.262723 0.000000 0.000000 0.00109 -0.00000 0.00000 MD: 5 H -2.348329 1.753459 0.000000 0.00124 -0.00063 0.00000 MD: 6 H -2.348329 -1.753459 0.000000 0.00124 0.00063 -0.00000 MD: 2 MD: 1 C 1.259460 0.000000 0.000001 0.00200 -0.00000 -0.00000 MD: 2 H 2.344603 1.751556 0.000000 -0.00085 -0.00008 0.00000 MD: 3 H 2.344603 -1.751556 -0.000000 -0.00085 0.00008 0.00000 MD: 4 C -1.259456 -0.000000 0.000000 -0.00200 0.00000 -0.00000 MD: 5 H -2.344603 1.751557 0.000000 0.00085 -0.00008 0.00000 MD: 6 H -2.344603 -1.751557 -0.000000 0.00085 0.00008 0.00000 MD: 3 MD: 1 C 1.262349 0.000000 0.000000 -0.00219 0.00000 0.00000 MD: 2 H 2.343376 1.751441 0.000000 -0.00034 0.00046 0.00000 MD: 3 H 2.343376 -1.751441 0.000000 -0.00034 -0.00046 0.00000 MD: 4 C -1.262347 0.000000 0.000000 0.00219 -0.00000 0.00000 MD: 5 H -2.343375 1.751441 0.000000 0.00034 0.00046 0.00000 MD: 6 H -2.343374 -1.751441 -0.000000 0.00034 -0.00046 0.00000 MD: 4 MD: 1 C 1.260579 -0.000000 -0.000000 0.00002 0.00000 0.00000 MD: 2 H 2.341738 1.752212 0.000000 -0.00040 0.00022 -0.00000 MD: 3 H 2.341738 -1.752212 0.000000 -0.00040 -0.00022 -0.00000 MD: 4 C -1.260578 0.000000 -0.000000 -0.00002 -0.00000 -0.00000 MD: 5 H -2.341735 1.752211 0.000000 0.00040 0.00022 -0.00000 MD: 6 H -2.341735 -1.752211 -0.000000 0.00040 -0.00022 -0.00000
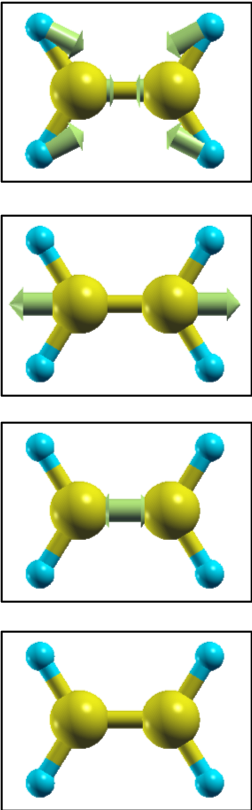
The maximal force acting on the atoms can be seen by extracting lines below those including f_max from nfout_1:
$ grep -A1 f_max nfout_1
NIT TotalEnergy f_max f_rms edel vdel fdel
1 -13.90231646 0.001395 0.001300 0.32D-09 0.80D-07 0.32D-09
--
NIT TotalEnergy f_max f_rms edel vdel fdel
2 -13.90233259 0.002005 0.001351 0.44D-10 0.45D-07 0.44D-10
--
NIT TotalEnergy f_max f_rms edel vdel fdel
3 -13.90233634 0.002190 0.001348 0.11D-08 0.13D-06 0.11D-08
--
NIT TotalEnergy f_max f_rms edel vdel fdel
4 -13.90234327 0.000451 0.000368 0.17D-08 0.14D-06 0.17D-08
We here learn how to find stable atomic adsorption sites on a metallic surface and perform vibrational mode analyses
A chlorine atom is adsorbed at the top site,
the bridge site,
and the hollow site.
Note that the atoms in the unit cell represented by a rectangular box is repeated infinitely.
$ cd ../ClonAl100 $ ls bridge hollow path top util
We optimize the adsoption structure of a chlorine atom on the top, bridge and hollow sites of the Al(100) surface in the corresponding directories. For example, the input file for the top-site adsorption reads:
$ cd top $ cat nfinp_1
1 0 0 0 0 0
4.00 10.00 2 7 7
1 0
CARTESIAN
7.6534000000 0.0000000000 0.0000000000
0.0000000000 7.6534000000 0.0000000000
0.0000000000 0.0000000000 30.6136000000
4 4 1 2 2 1
1 0
0.0000000000 3.8267000000 4.0000000000 1 1 2
0.0000000000 3.8267000000 0.0000000000 1 1 1
3.8267000000 0.0000000000 0.0000000000 1 1 1
0.0000000000 0.0000000000 -3.8267000000 1 0 1
3.8267000000 3.8267000000 -3.8267000000 1 0 1
0.0000000000 3.8267000000 -7.6534000000 1 0 1
3.8267000000 0.0000000000 -7.6534000000 1 0 1
13 0.15 26.9815 3 1 0.00
17 0.15 35.4527 3 1 0.00
0 0 0 0 0
0 1
100 200 0 57200.00 0
3 1
0 20 0.8
0.60 0.50 0.60 0.70 1.00
600.00 4 1 0.10D-08
-0.002 0.10D-02 0
ggapbe 1
1.00
101
0 0 0
0 0 0
16
1
0
2
0
0
0 0.00
By comparing the input files in top, bridge and hollow directories,
you can confirm that only the coordinates of the Cl atom different among these files,
correspondig to respective adsorption sites.
$ qsub qsub1.sh
Your job 1270 ("Al100-Cl") has been submitted
$ cd ../bridge
$ qsub qsub1.sh
Your job 1271 ("Al100-Cl") has been submitted
$ cd ../hollow
$ qsub qsub1.sh
Your job 1272 ("Al100-Cl") has been submitted
For each adsorption site, the structure of Cl on Al(100) is optimized with the lower two Al layers fixed.
$ cd ..
$ grep -A1 f_max top/enfout_1
NIT TotalEnergy f_max f_rms edel vdel fdel
1 -27.41198802 0.017836 0.015245 0.47D-09 0.52D-07 0.15D-09
--
NIT TotalEnergy f_max f_rms edel vdel fdel
2 -27.41294563 0.009333 0.008059 0.17D-08 0.19D-07 0.73D-10
--
NIT TotalEnergy f_max f_rms edel vdel fdel
3 -27.41345545 0.005091 0.003227 0.84D-09 0.83D-08 0.49D-11
--
NIT TotalEnergy f_max f_rms edel vdel fdel
4 -27.41355911 0.008506 0.006727 0.40D-09 0.22D-07 0.43D-09
--
NIT TotalEnergy f_max f_rms edel vdel fdel
5 -27.41336440 0.011139 0.008878 0.65D-10 0.10D-07 0.17D-10
--
NIT TotalEnergy f_max f_rms edel vdel fdel
6 -27.41141149 0.026651 0.021697 0.66D-10 0.22D-07 0.73D-10
--
NIT TotalEnergy f_max f_rms edel vdel fdel
7 -27.41382388 0.002234 0.001695 0.99D-11 0.19D-07 0.25D-12
--
NIT TotalEnergy f_max f_rms edel vdel fdel
8 -27.41235320 0.015765 0.012354 0.53D-09 0.36D-07 0.71D-10
--
NIT TotalEnergy f_max f_rms edel vdel fdel
9 -27.41367010 0.006512 0.005269 0.30D-10 0.35D-07 0.66D-10
--
NIT TotalEnergy f_max f_rms edel vdel fdel
10 -27.41381140 0.002926 0.002341 0.12D-10 0.16D-07 0.53D-10
--
NIT TotalEnergy f_max f_rms edel vdel fdel
11 -27.41384468 0.000625 0.000441 0.13D-09 0.34D-07 0.17D-09
$ grep -A1 f_max bridge/nfout_1
NIT TotalEnergy f_max f_rms edel vdel fdel
1 -27.41551837 0.008329 0.006823 0.13D-08 0.61D-08 0.42D-10
--
NIT TotalEnergy f_max f_rms edel vdel fdel
2 -27.41630364 0.005081 0.004658 0.69D-10 0.78D-08 0.23D-10
--
NIT TotalEnergy f_max f_rms edel vdel fdel
3 -27.41679245 0.003830 0.002644 0.58D-09 0.99D-08 0.13D-10
--
NIT TotalEnergy f_max f_rms edel vdel fdel
4 -27.41696629 0.001321 0.001181 0.16D-08 0.15D-07 0.22D-11
--
NIT TotalEnergy f_max f_rms edel vdel fdel
5 -27.41575934 0.007502 0.006209 0.25D-08 0.72D-08 0.13D-11
--
NIT TotalEnergy f_max f_rms edel vdel fdel
6 -27.41698675 0.001387 0.001139 0.37D-09 0.15D-07 0.15D-10
--
NIT TotalEnergy f_max f_rms edel vdel fdel
7 -27.41702702 0.000476 0.000390 0.52D-09 0.33D-08 0.33D-10
grep -A1 f_max hollow/nfout_1
NIT TotalEnergy f_max f_rms edel vdel fdel
1 -27.39937930 0.003518 0.002925 0.29D-09 0.51D-07 0.16D-09
--
NIT TotalEnergy f_max f_rms edel vdel fdel
2 -27.39950767 0.002422 0.001986 0.25D-08 0.13D-06 0.15D-08
--
NIT TotalEnergy f_max f_rms edel vdel fdel
3 -27.39958878 0.001534 0.001099 0.15D-08 0.40D-08 0.14D-10
--
NIT TotalEnergy f_max f_rms edel vdel fdel
4 -27.39963087 0.000666 0.000575 0.27D-08 0.16D-07 0.39D-10
We next investigate the vibrational modes using the optmized electronic states (ICOND = 1)
and surface structures,
whose coordinates are already reflected in the second input files:
$ cd top $ cat nfinp_2
1 0 0 0 0 0
4.000 10.0000 2 7 7
1 0
CARTESIAN
7.6534000000 0.0000000000 00.0000000000
0.0000000000 7.6534000000 00.0000000000
00.0000000000 00.0000000000 30.6136000000
4 4 1 2 2 1
1 0
-0.000000251836 3.826701578025 4.049619608896 1 1 2
-0.000001836163 3.826699462952 -0.045772180307 1 1 1
3.826699127649 0.000000430959 -0.233606043422 1 1 1
0.000000000000 0.000000000000 -3.826700000000 1 0 1
3.826700000000 3.826700000000 -3.826700000000 1 0 1
0.000000000000 3.826700000000 -7.653400000000 1 0 1
3.826700000000 0.000000000000 -7.653400000000 1 0 1
13 0.15 26.98150000 3 1 0.00000000
17 0.15 35.45270000 3 1 0.00000000
1 0 0 0 0
0 1
100 200 0 57200.00 0
3 1
0 20 0.8
0.60 0.50 0.60 0.70 1.00
600.00 3 1 0.10D-08 1.d-06
-0.0020 0.10D-02 0
ggapbe 1
1.00 3
102
0 0 0
0 0 0
16
1
0
2 0 0 0(MB)
0
0
0 0.0
In addition to the coordinates of the surface atoms,
IMDALG is also changed to 3:
| Parameter | Value | Description |
|---|---|---|
ICOND |
1 |
Restart calculation using old wave functinos and charge density |
IMDALG |
3 |
Vibrational mode analysis |
$ cat nfvibrate.data
1 1.0d0 1 0.0000000000 0.0000000000 0.000000000000 1 1.0d0 1 0.1000000000 0.0000000000 0.000000000000 1 -1.0d0 1 0.1000000000 0.0000000000 0.000000000000 1 1.0d0 1 0.0000000000 0.1000000000 0.000000000000 1 -1.0d0 1 0.0000000000 0.1000000000 0.000000000000 1 1.0d0 1 0.0000000000 0.0000000000 0.100000000000 1 -1.0d0 1 0.0000000000 0.0000000000 0.100000000000
$ qsub qsub2.sh
Your job 1273 ("Al100-Cl") has been submitted
$ grep MD: nfout_2 | tr -d "[MD:ACl]" > MD_2.out $ cat MD_2.out
1
1 -0.000000 3.826702 4.049620 0.00000 0.00000 0.00086
2 -0.000002 3.826699 -0.045772 0.00000 0.00000 -0.00042
3 3.826699 0.000000 -0.233606 0.00000 -0.00000 0.00022
4 0.000000 0.000000 -3.826700 -0.00000 -0.00000 0.00031
5 3.826700 3.826700 -3.826700 -0.00000 0.00000 0.00031
6 0.000000 3.826700 -7.653400 0.00000 0.00000 -0.00155
7 3.826700 0.000000 -7.653400 -0.00000 -0.00000 0.00029
2
1 0.100000 3.826702 4.049620 -0.00005 -0.00000 0.00075
2 -0.000002 3.826699 -0.045772 -0.00043 0.00000 -0.00031
3 3.826699 0.000000 -0.233606 0.00033 -0.00000 0.00024
4 0.000000 0.000000 -3.826700 0.00021 -0.00000 0.00030
5 3.826700 3.826700 -3.826700 -0.00005 0.00000 0.00031
6 0.000000 3.826700 -7.653400 0.00020 0.00000 -0.00156
7 3.826700 0.000000 -7.653400 -0.00021 -0.00000 0.00028
3
1 -0.100000 3.826702 4.049620 0.00005 0.00000 0.00076
2 -0.000002 3.826699 -0.045772 0.00043 0.00000 -0.00031
3 3.826699 0.000000 -0.233606 -0.00033 -0.00000 0.00025
4 0.000000 0.000000 -3.826700 -0.00021 -0.00000 0.00030
5 3.826700 3.826700 -3.826700 0.00005 0.00000 0.00031
6 0.000000 3.826700 -7.653400 -0.00020 0.00000 -0.00156
7 3.826700 0.000000 -7.653400 0.00021 -0.00000 0.00028
4
1 -0.000000 3.926702 4.049620 -0.00000 -0.00005 0.00076
2 -0.000002 3.826699 -0.045772 0.00000 -0.00043 -0.00031
3 3.826699 0.000000 -0.233606 0.00000 0.00033 0.00024
4 0.000000 0.000000 -3.826700 -0.00000 -0.00005 0.00031
5 3.826700 3.826700 -3.826700 -0.00000 0.00021 0.00030
6 0.000000 3.826700 -7.653400 0.00000 0.00020 -0.00156
7 3.826700 0.000000 -7.653400 -0.00000 -0.00021 0.00028
5
1 -0.000000 3.726702 4.049620 -0.00000 0.00005 0.00076
2 -0.000002 3.826699 -0.045772 0.00000 0.00043 -0.00031
3 3.826699 0.000000 -0.233606 0.00000 -0.00033 0.00025
4 0.000000 0.000000 -3.826700 -0.00000 0.00005 0.00031
5 3.826700 3.826700 -3.826700 -0.00000 -0.00021 0.00030
6 0.000000 3.826700 -7.653400 0.00000 -0.00020 -0.00156
7 3.826700 0.000000 -7.653400 -0.00000 0.00021 0.00028
6
1 -0.000000 3.826702 4.149620 0.00000 -0.00000 -0.01065
2 -0.000002 3.826699 -0.045772 0.00000 0.00000 0.01014
3 3.826699 0.000000 -0.233606 0.00000 -0.00000 0.00138
4 0.000000 0.000000 -3.826700 -0.00000 -0.00000 0.00016
5 3.826700 3.826700 -3.826700 -0.00000 0.00000 0.00016
6 0.000000 3.826700 -7.653400 0.00000 0.00000 -0.00146
7 3.826700 0.000000 -7.653400 -0.00000 -0.00000 0.00031
7
1 -0.000000 3.826702 3.949620 0.00000 0.00000 0.01534
2 -0.000002 3.826699 -0.045772 0.00000 0.00000 -0.01392
3 3.826699 0.000000 -0.233606 0.00000 -0.00000 -0.00096
4 0.000000 0.000000 -3.826700 -0.00000 -0.00000 0.00046
5 3.826700 3.826700 -3.826700 -0.00000 0.00000 0.00046
6 0.000000 3.826700 -7.653400 0.00000 0.00000 -0.00165
7 3.826700 0.000000 -7.653400 -0.00000 -0.00000 0.00025
The input file for the vibrational mode analysis MD_2.data is made by removing
the first block of displacement and adding the first two lines:
7 3
36 27 27 27 27 27 27
2
1 0.100000 3.826702 4.049620 -0.00005 -0.00000 0.00075
2 -0.000002 3.826699 -0.045772 -0.00043 0.00000 -0.00031
3 3.826699 0.000000 -0.233606 0.00033 -0.00000 0.00024
4 0.000000 0.000000 -3.826700 0.00021 -0.00000 0.00030
5 3.826700 3.826700 -3.826700 -0.00005 0.00000 0.00031
6 0.000000 3.826700 -7.653400 0.00020 0.00000 -0.00156
7 3.826700 0.000000 -7.653400 -0.00021 -0.00000 0.00028
3
1 -0.100000 3.826702 4.049620 0.00005 0.00000 0.00076
2 -0.000002 3.826699 -0.045772 0.00043 0.00000 -0.00031
3 3.826699 0.000000 -0.233606 -0.00033 -0.00000 0.00025
4 0.000000 0.000000 -3.826700 -0.00021 -0.00000 0.00030
5 3.826700 3.826700 -3.826700 0.00005 0.00000 0.00031
6 0.000000 3.826700 -7.653400 -0.00020 0.00000 -0.00156
7 3.826700 0.000000 -7.653400 0.00021 -0.00000 0.00028
4
1 -0.000000 3.926702 4.049620 -0.00000 -0.00005 0.00076
2 -0.000002 3.826699 -0.045772 0.00000 -0.00043 -0.00031
3 3.826699 0.000000 -0.233606 -0.00000 0.00033 0.00024
4 0.000000 0.000000 -3.826700 -0.00000 -0.00005 0.00031
5 3.826700 3.826700 -3.826700 -0.00000 0.00021 0.00030
6 0.000000 3.826700 -7.653400 0.00000 0.00020 -0.00156
7 3.826700 0.000000 -7.653400 -0.00000 -0.00021 0.00028
5
1 -0.000000 3.726702 4.049620 0.00000 0.00005 0.00076
2 -0.000002 3.826699 -0.045772 0.00000 0.00043 -0.00031
3 3.826699 0.000000 -0.233606 -0.00000 -0.00033 0.00025
4 0.000000 0.000000 -3.826700 -0.00000 0.00005 0.00031
5 3.826700 3.826700 -3.826700 -0.00000 -0.00021 0.00030
6 0.000000 3.826700 -7.653400 0.00000 -0.00020 -0.00156
7 3.826700 0.000000 -7.653400 -0.00000 0.00021 0.00028
6
1 -0.000000 3.826702 4.149620 0.00000 -0.00000 -0.01065
2 -0.000002 3.826699 -0.045772 0.00000 0.00000 0.01014
3 3.826699 0.000000 -0.233606 0.00000 -0.00000 0.00138
4 0.000000 0.000000 -3.826700 -0.00000 -0.00000 0.00016
5 3.826700 3.826700 -3.826700 -0.00000 0.00000 0.00016
6 0.000000 3.826700 -7.653400 0.00000 0.00000 -0.00146
7 3.826700 0.000000 -7.653400 -0.00000 -0.00000 0.00031
7
1 -0.000000 3.826702 3.949620 0.00000 0.00000 0.01534
2 -0.000002 3.826699 -0.045772 0.00000 0.00000 -0.01392
3 3.826699 0.000000 -0.233606 0.00000 -0.00000 -0.00096
4 0.000000 0.000000 -3.826700 -0.00000 -0.00000 0.00046
5 3.826700 3.826700 -3.826700 -0.00000 0.00000 0.00046
6 0.000000 3.826700 -7.653400 0.00000 0.00000 -0.00165
7 3.826700 0.000000 -7.653400 -0.00000 -0.00000 0.00025
In MD_2,data, the first line specifies the numbers of atoms and
vibration modes, respectively,
and the second line specifies the atomic masses in units of
the hydrogen mass.
Finally, the vibrational modes and the corresponding zero-point energies
are calculated using program gif in directory ~/STATE/util/Vibration
$ ~/STATE/util/Vibration/src/gif < MD_2.data
.
.
.
Vibrational frequencies and eigen vectors
NATM FAC MODE WR : NU(meV) NU(cm-1)
7 0.10D+01 1 0.14D-04 : 2.37 19.09
1 0.1666666636 0.0000000000 -0.0000321875
2 0.0000000000 0.0000000000 0.0000000000
3 0.0000000000 0.0000000000 0.0000000000
4 0.0000000000 0.0000000000 0.0000000000
5 0.0000000000 0.0000000000 0.0000000000
6 0.0000000000 0.0000000000 0.0000000000
7 0.0000000000 0.0000000000 0.0000000000
7 0.10D+01 2 0.14D-04 : 2.37 19.09
1 0.0000000000 -0.1666666667 -0.0000000000
2 0.0000000000 0.0000000000 0.0000000000
3 0.0000000000 0.0000000000 0.0000000000
4 0.0000000000 0.0000000000 0.0000000000
5 0.0000000000 0.0000000000 0.0000000000
6 0.0000000000 0.0000000000 0.0000000000
7 0.0000000000 0.0000000000 0.0000000000
7 0.15D+00 3 0.36D-02 : 38.15 307.73
1 0.0000321875 -0.0000000000 0.1666666636
2 0.0000000000 0.0000000000 0.0000000000
3 0.0000000000 0.0000000000 0.0000000000
4 0.0000000000 0.0000000000 0.0000000000
5 0.0000000000 0.0000000000 0.0000000000
6 0.0000000000 0.0000000000 0.0000000000
7 0.0000000000 0.0000000000 0.0000000000
=======
SUMMARY
=======
MODE WR : NU(meV) NU(cm-1)
1 0.14D-04 : 2.37 19.09
2 0.14D-04 : 2.37 19.09
3 0.36D-02 : 38.15 307.73
Similar analyses can be done in directories bridge
.
.
.
Vibrational frequencies and eigen vectors
NATM FAC MODE WR : NU(meV) NU(cm-1)
7 0.10D+01 1 0.15D-03 : 7.78 62.73
1 0.1178489705 -0.1178532500 -0.0000970020
2 0.0000000000 0.0000000000 0.0000000000
3 0.0000000000 0.0000000000 0.0000000000
4 0.0000000000 0.0000000000 0.0000000000
5 0.0000000000 0.0000000000 0.0000000000
6 0.0000000000 0.0000000000 0.0000000000
7 0.0000000000 0.0000000000 0.0000000000
7 0.10D+01 2 0.24D-03 : 9.93 80.08
1 0.1178531952 0.1178486620 0.0003082383
2 0.0000000000 0.0000000000 0.0000000000
3 0.0000000000 0.0000000000 0.0000000000
4 0.0000000000 0.0000000000 0.0000000000
5 0.0000000000 0.0000000000 0.0000000000
6 0.0000000000 0.0000000000 0.0000000000
7 0.0000000000 0.0000000000 0.0000000000
7 0.30D+00 3 0.18D-02 : 27.22 219.56
1 -0.0001493719 -0.0002865454 0.1666663534
2 0.0000000000 0.0000000000 0.0000000000
3 0.0000000000 0.0000000000 0.0000000000
4 0.0000000000 0.0000000000 0.0000000000
5 0.0000000000 0.0000000000 0.0000000000
6 0.0000000000 0.0000000000 0.0000000000
7 0.0000000000 0.0000000000 0.0000000000
=======
SUMMARY
=======
MODE WR : NU(meV) NU(cm-1)
1 0.15D-03 : 7.78 62.73
2 0.24D-03 : 9.93 80.08
3 0.18D-02 : 27.22 219.56
and hollow:
.
.
.
Vibrational frequencies and eigen vectors
NATM FAC MODE WR : NU(meV) NU(cm-1)
7 0.10D+01 1 -0.36D-03 : 12.02 96.96
1 -0.1178511111 0.1178511111 0.0000949646
2 0.0000000000 0.0000000000 0.0000000000
3 0.0000000000 0.0000000000 0.0000000000
4 0.0000000000 0.0000000000 0.0000000000
5 0.0000000000 0.0000000000 0.0000000000
6 0.0000000000 0.0000000000 0.0000000000
7 0.0000000000 0.0000000000 0.0000000000
7 0.10D+01 2 -0.36D-03 : 12.02 96.96
1 0.1178511302 0.1178511302 0.0000000000
2 0.0000000000 0.0000000000 0.0000000000
3 0.0000000000 0.0000000000 0.0000000000
4 0.0000000000 0.0000000000 0.0000000000
5 0.0000000000 0.0000000000 0.0000000000
6 0.0000000000 0.0000000000 0.0000000000
7 0.0000000000 0.0000000000 0.0000000000
7 0.41D+00 3 0.14D-02 : 23.46 189.25
1 0.0000671501 -0.0000671501 0.1666666396
2 0.0000000000 0.0000000000 0.0000000000
3 0.0000000000 0.0000000000 0.0000000000
4 0.0000000000 0.0000000000 0.0000000000
5 0.0000000000 0.0000000000 0.0000000000
6 0.0000000000 0.0000000000 0.0000000000
7 0.0000000000 0.0000000000 0.0000000000
=======
SUMMARY
=======
MODE WR : NU(meV) NU(cm-1)
1 -0.36D-03 : 12.02 96.96
2 -0.36D-03 : 12.02 96.96
3 0.14D-02 : 23.46 189.25